Lipoprotein(a), or Lp(a), is a genetically determined LDL-like particle that increases the risk of atherosclerotic cardiovascular disease and calcific aortic valve stenosis. Because Lp(a) is not included in a standard lipid panel and is only minimally affected by lifestyle, it can remain hidden unless specifically measured. Despite affecting roughly one in five people worldwide, Lp(a) remains undertested in routine practice. Current guidance recommends measuring Lp(a) at least once in adulthood, because an elevated level can change how aggressively LDL-C and other modifiable cardiovascular risk factors should be managed.
- Measure Lp(a) at least once in adulthood
- Consider ≥125 nmol/L (≥50 mg/dL) a high-risk threshold
- Use Lp(a) as a risk-enhancing factor, not a routine treatment target
- Pair Lp(a) with PREVENT, CAC, LDL-C, and sometimes ApoB for better risk assessment
- Phase 3 outcome trials for pelacarsen, olpasiran and lepodisiran are expected to report from 2026
For a plain-English summary, see our patient guide to high Lp(a). For the broader guideline framework, see our 2026 dyslipidemia guideline summary.
Why Lp(a) matters clinically
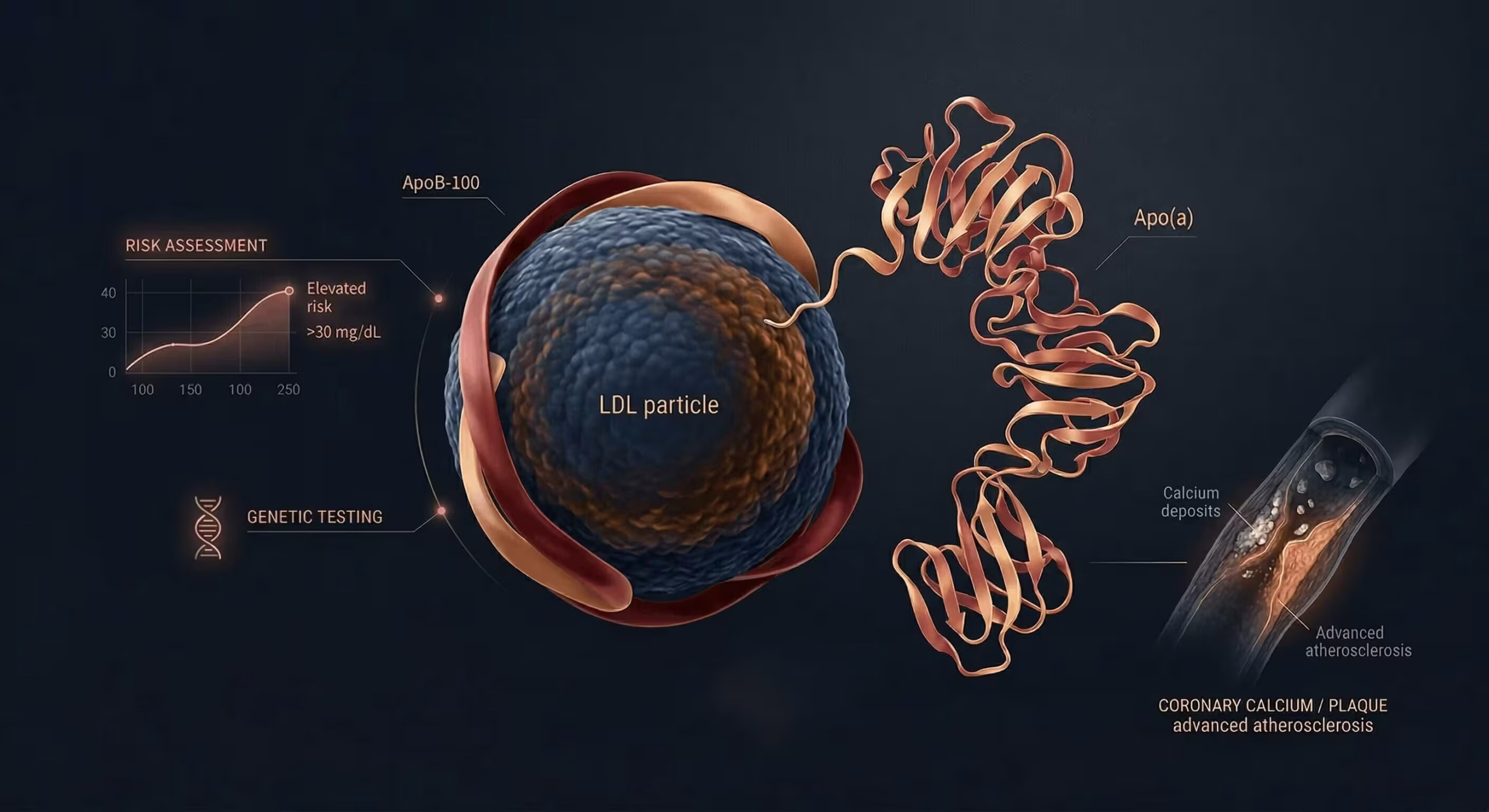
Lp(a) captures inherited cardiovascular risk that is often missed by the standard lipid panel. High Lp(a) is associated with coronary artery disease, ischaemic stroke, peripheral artery disease, and calcific aortic valve stenosis. Importantly, patients with apparently well-controlled LDL-C may still carry substantial residual risk if Lp(a) is markedly elevated. On a per-particle basis, an Lp(a) particle is estimated to be approximately six-fold more atherogenic than an LDL particle (Björnson et al., JACC 2024).
This is one of the reasons Lp(a) has moved from a niche specialist test to a mainstream part of cardiovascular risk assessment. For many patients, the value of the result is not that it changes one lab number, but that it changes how seriously overall risk is taken and how intensive preventive therapy becomes. The 2026 ACC/AHA dyslipidemia guideline now explicitly includes Lp(a) as a risk-enhancing factor that can influence treatment decisions.
Who should be tested for Lp(a)?
Routine testing
Current cardiovascular guidance — including from the National Lipid Association and the 2026 ACC/AHA guideline — recommends measuring Lp(a) at least once in adulthood. Because levels are largely genetically determined and usually stable over time, a single measurement is enough for most people (Class of Recommendation I, Level of Evidence B-NR).
When testing is especially helpful
Lp(a) testing is particularly useful in people with premature cardiovascular disease (before age 55 in men, 65 in women), a strong family history of early heart disease, known or suspected familial hypercholesterolaemia, unexplained residual risk despite well-controlled LDL-C, borderline or intermediate overall risk where treatment decisions are uncertain, or calcific aortic valve disease. In children, selective screening is recommended when there is a family history of FH, premature ASCVD, or a first-degree relative with elevated Lp(a). Our FH calculator can help structure the assessment for inherited lipid disorders.
How should Lp(a) be measured and interpreted?
Preferred units
Lp(a) is best interpreted in the units reported by the laboratory. When available, nmol/L is generally preferred because it reflects particle number more directly and is less distorted by apolipoprotein(a) size variability than mg/dL. Immunochemical assays calibrated against the WHO/IFCCLM reference material are the recommended method.
Why mg/dL and nmol/L cannot be reliably converted
Unlike standard cholesterol measures, Lp(a) does not have a dependable fixed conversion factor between mg/dL and nmol/L. The apo(a) component varies substantially in size due to kringle IV type 2 (KIV-2) repeat polymorphism — from 3 to over 40 copies — so the same mass concentration may represent different particle numbers. For this reason, results should be interpreted in the units provided by the assay.
Practical interpretation of levels
(<30 mg/dL)
(30–50 mg/dL)
(≥50 mg/dL)
(~100 mg/dL)
Risk is continuous, not all-or-none. A value just below a threshold is not "normal" in an absolute sense, and a very high value matters more in someone who also has elevated LDL-C, diabetes, hypertension, smoking, or coronary calcium. Levels around 250 nmol/L are associated with roughly double the long-term risk of cardiovascular events. This is where the next section — integrating Lp(a) with other risk tools — becomes particularly useful.
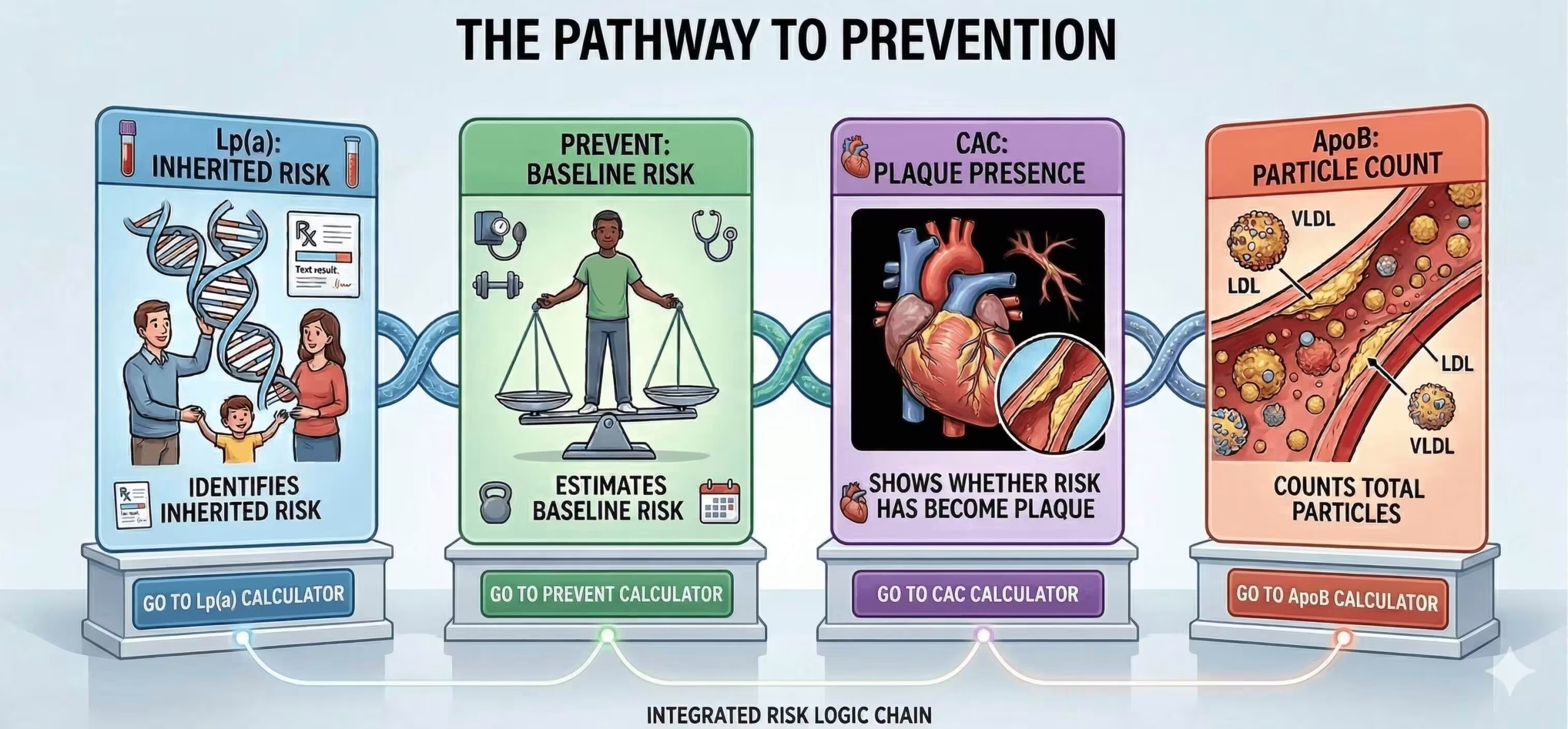
How elevated Lp(a) changes risk assessment: PREVENT, CAC and ApoB
An elevated Lp(a) result should not be viewed in isolation. Its real value is in changing the interpretation of overall cardiovascular risk — especially in patients whose risk estimate is borderline, whose LDL-C appears reasonable, or whose family history suggests more risk than the standard lipid panel shows.
PREVENT
The PREVENT equations are now the preferred tool for primary prevention risk estimation in adults aged 30–79 years. Lp(a) is not a replacement for PREVENT; rather, it adds context to the estimated risk and may justify a lower threshold for intensifying prevention when inherited risk is clearly elevated.
Coronary artery calcium (CAC)
If treatment decisions remain uncertain after standard risk assessment, coronary artery calcium scoring can help reclassify risk. In the 2026 dyslipidemia guideline, selective CAC use is recommended in borderline or intermediate-risk patients, and the result can influence LDL-C goals and treatment intensity. In practice, Lp(a) and CAC work well together: Lp(a) identifies inherited risk, while CAC shows whether that risk has already translated into measurable plaque burden. For more on how calcium scoring is used clinically, see our dedicated article.
ApoB
Lp(a) and ApoB are related, but they are not interchangeable. Each Lp(a) particle carries one ApoB-100 molecule, but ApoB reflects the total number of atherogenic particles across LDL, IDL, VLDL remnants, and Lp(a). That makes ApoB especially useful when LDL-C appears acceptable but residual risk remains high — for example in diabetes, metabolic syndrome, hypertriglyceridaemia, chronic kidney disease, or established cardiovascular disease. Where LDL-C and ApoB are discordant, ApoB is generally the better guide to atherogenic burden.
Management of elevated Lp(a)
A high Lp(a) result usually does not mean there is a specific Lp(a)-targeted treatment to start today. Instead, it means the patient's overall cardiovascular risk should be taken more seriously. In most cases, the practical response is to lower LDL-C more intensively, treat blood pressure and diabetes carefully, stop smoking, and consider additional risk refinement with ApoB or CAC if management remains uncertain.
LDL-C lowering: statins, ezetimibe and PCSK9 inhibitors
Statins do not directly lower Lp(a) — and may cause a modest increase — but they still reduce cardiovascular risk and remain foundational therapy when LDL-C lowering is indicated. Ezetimibe may be added when LDL-C goals are not achieved with statins alone. PCSK9 inhibitors produce a modest Lp(a) reduction of approximately 20–30% in addition to their potent LDL-C lowering, and may be considered in high-risk patients who do not reach target on maximally tolerated statin plus ezetimibe. For patients with familial hypercholesterolaemia, PCSK9-based therapy may be particularly important when both LDL-C and Lp(a) are elevated.
Lipoprotein apheresis
Apheresis remains a niche but important option in selected very high-risk patients — particularly those with FH, established ASCVD or PAD, Lp(a) ≥60 mg/dL (~150 nmol/L), and LDL-C ≥100 mg/dL despite maximally tolerated therapy. Apheresis can lower both LDL-C and Lp(a) by 60–70% acutely, with a time-averaged Lp(a) reduction of approximately 30–35%.
Repeat testing and family screening
Because Lp(a) is largely genetic and stable, repeat testing is usually unnecessary unless there is a specific clinical reason (such as changes in renal function or menopause). An elevated result should prompt consideration of cascade screening in first-degree relatives, especially when there is a family history of premature ASCVD or familial hypercholesterolaemia.
Aspirin
Aspirin should not be presented as routine Lp(a)-directed therapy. If considered, it should be framed within standard primary or secondary prevention decision-making, weighing cardiovascular benefit against individual bleeding risk.
Emerging Lp(a)-lowering therapies
Targeted Lp(a) lowering is one of the most active areas in preventive cardiology. Several RNA-based therapies have demonstrated reductions of 80–94% in phase 2 trials, and major phase 3 cardiovascular outcome trials are now underway. Definitive event-reduction data are still pending — meaning Lp(a) is already clinically important today, but routine treatment still focuses on LDL-C and overall risk reduction until outcome trials report.
Trial: Lp(a)HORIZON · Phase 3 · n=8,323
Results expected: H1 2026
Phase 2: ~80% Lp(a) reduction
Dosing: 80 mg SC monthly
Trial: OCEAN(a) · Phase 3 · n>7,200
Results expected: Late 2026–2027
Phase 2: ~94% Lp(a) reduction
Dosing: SC every 12 weeks
Trial: ACCLAIM-Lp(a) · Phase 3 · n≈12,500
Results expected: ~2029
Phase 2: 94% reduction at 180 days (single dose)
Dosing: SC once or twice yearly
Trial: KRAKEN · Phase 2 completed
Status: Phase 3 planning
Phase 2: Up to 86% Lp(a) reduction
Dosing: Oral daily
